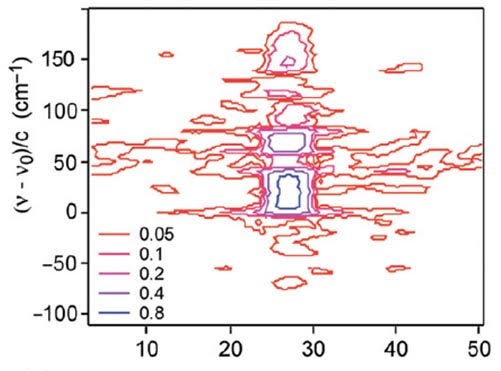
Quantum Information scrambling in molecules
Out-of-time-order correlators (OTOCs) can be used to probe how quickly a quantum system scrambles information when the initial conditions of the dynamics are changed. In sufficiently large quantum systems, one can extract from the OTOC the quantum analog of the Lyapunov coefficient that describes the timescale on which a classical chaotic system becomes scrambled. The vibrations of polyatomic molecules are known to undergo a transition from regular dynamics at low energy to facile energy flow at sufficiently high energy. Molecules therefore represent ideal quantum systems to study scrambling in many-body systems of moderate size. By computing quantum OTOCs and their classical counterparts we quantify how information becomes “scrambled” quantum mechanically in molecular systems. Between early “ballistic” dynamics, and late “saturation” of the OTOC when the full density of states is explored, there is indeed a regime where a quantum Lyapunov coefficient can be defined for all molecules in this study. Comparison with experimental rate data shows that slow scrambling as measured by the OTOC can reach the timescale of molecular reaction dynamics.
C. Zhang, P. G. Wolynes, and M. Gruebele, Quantum Information Scrambling in Molecules, Phys. Rev. A 105, 033322 (2022).
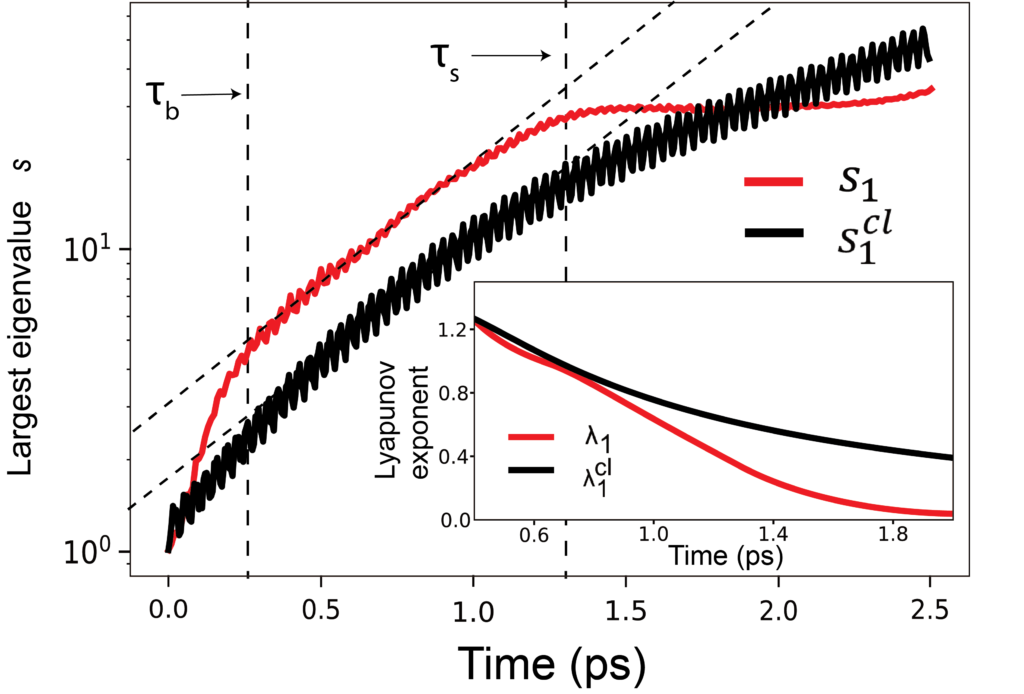
Energy flow limited reactivity
Intramolecular energy flow is often assumed im RRKM, transition state and other rate theory not to be an impediment to reaction. In contrast, evidence from spectroscopy , computational study and Anderson-localization based model have shown limited ergodicity during molecular energy flow.
We develop a simple model for the interplay of IVR and energy transfer as shown in the figure and simulate the model with near-exact quantum dynamics . A sharp ‘phase transition’ is observed as function of molecular anharmonicity between facile and limited IVR . This narrow transition range happens to lie right in the middle of the range expected for molecular torsion, bending, and stretching vibrations, suggesting reactive energy transfer dynamics not far from localization boundary, with implications for controllability of reactions.
Chenghao Zhang, Edwin L. Sibert III, and Martin Gruebele , “A phase diagram for energy flow-limited reactivity”, The Journal of Chemical Physics 154,104301 (2021) https://doi.org/10.1063/5.0043665
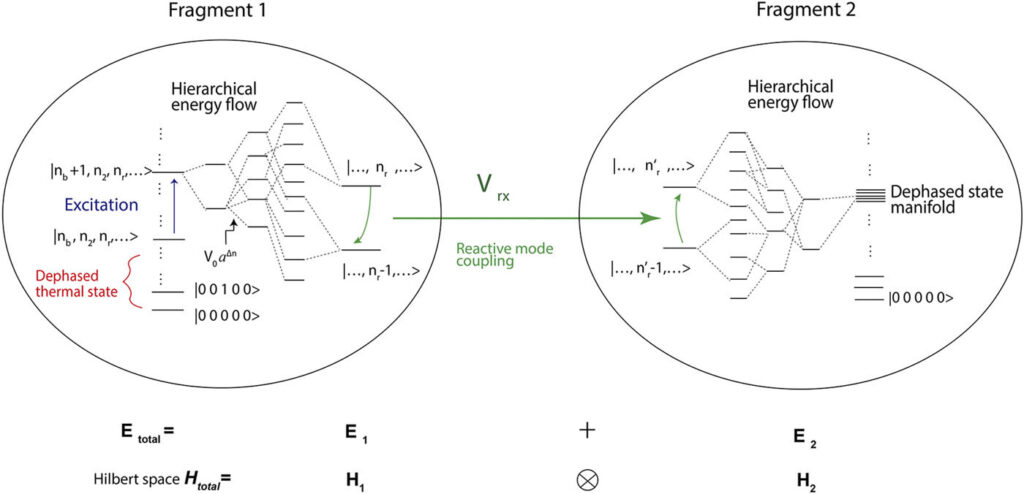
Transport through spatio-temporally inhomogeneous media
It is useful to have a systematic and simple framework to describe transport and equilibrium in chemical systems. We study Lagrangian-based approaches to describe transport phenomena, while remaining consistent with chemical thermodynamics. A model that neglects heat flow is suitable for the modern array of microscopy experiments that apply perturbations such as temperature or osmotic pressure jumps to inhomogeneous media such as living cells or polymer coatings.
The ease with which our approach derives transport and equilibrium relations for any type of driving factors (electromagnetic, thermal, pressure, surface tension, etc.) should make our model useful in physics, chemistry, biophysics, engineering, and even economics and social sciences.
N. Kocherginsky, and M. Gruebele, “A Mechanical Approach to Chemical Transport,” Proc. Natl. Acad. Sci USA, https://doi.org/10.1073/pnas.1600866113 (2016).
Molecular energy flow and its control
Energy flow is of fundamental importance as the initial step in chemical reactions. In essence, molecules roughly behave like a set of balls connected by anharmonic springs: pulling on one of these springs will soon cause the whole contraption to vibrate in an apparently random fashion.
Or is it as random as it seems? We investigate this problem theorectically for large and highly excited molecules, with tools ranging from accurate ab-initio based force fields to simple scaling laws based on underlying symmetries. We also study the problem experimentally by frequency- and time-resolved stimulated emission spectroscopy. We find that vibrational energy redistribution slows dramatically as the molecule explores more of the available quantum states. This opens up the possibility of contolling energy flow.
E. Berrios, and M. Gruebele, “Franck-Condon fingerprinting of vibration-tunneling spectra,” J. Phys. Chem. A 117, 7535-7541 (2013).
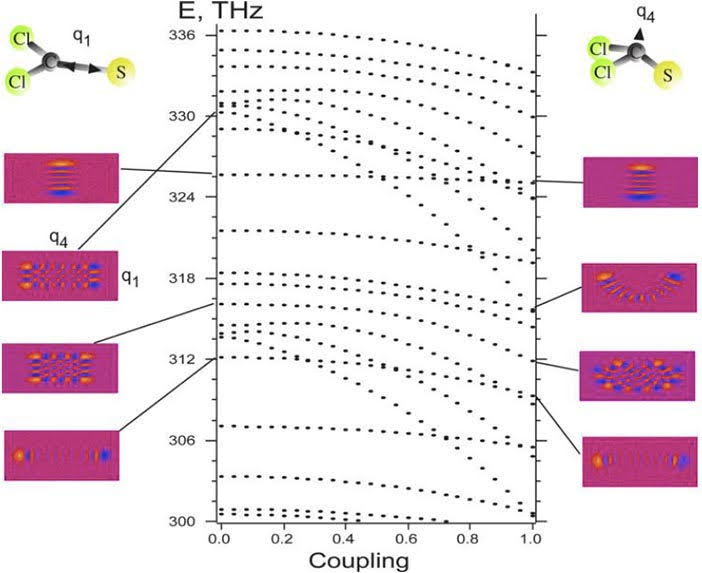
Molecular quantum computing
Qubits are quantum mechanical two-level systems that can be in state “0”, state “1”, or a superposition of the two at the same time. This is where they differ from classical bits, and offer the possibility of simulating NP-hard problems, improved encyption, and so forth.
We study how such qubits could be implemented and read out in molecules, where vibrational or rotational dynamics offers mutilevel systems in which qubits can be multiplexed. A single molecule can hold many more qubits than a two level spin or atomic system. The figure shows implementation and fidelity of Shor’s algorithm for factoring numbers using femtosecond pulses and the molecule SCCl2.
D. Shyshlov, E. Berrios, M. Gruebele, D. Babikov and “On readout of vibrational qubits using quantum beats”, J. Chem. Phys. 141, 224306 (1-7) (2014).